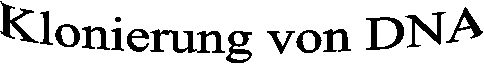
Teil 1: Durchführung
Versuchsauswertung: Schneiden des Plasmids pUCD mit
Restriktionsenzymen
Versuchsauswertung: Transformation und Klonierung
Diskutieren der Versuchsergebnisse und Fazit :
Bitte benutzen Sie die obigen Links zur Navigation, um zu Ihrer gewünschten Stelle zu springen, oder lesen Sie das ganze Protokoll indem Sie einfach runterscrollen.
Nachdem wir im vorangegangenen Versuch erfahren haben wie man DNA analysiert, beginnen wir mit Versuch C. Hier geht es im Wesentlichen darum, DNA zu klonieren. Dies bedeutet, dass man eine fremde DNA auf experimentellem Wege in einen Organismus überträgt. Diese fremde DNA muss von dem Organismus eingebaut, abgelesen und verdoppelt werden.
Für den Versuch werden E. coli Bakterien eingesetzt, da mit deren Anwendung gentechnische Methoden einfach demonstriert werden können.
Die einzelnen Schritte umfassen zunächst die Bearbeitung der DNA, die Transformation,
d. h. das Einschleusen der veränderten DNA in andere Bakterienzellen und schließlich die Selektion, d. h. das Auffinden und Isolieren der gentechnisch modifizierten Bakterien. Diesen gesamten Vorgang bezeichnet man auch als Klonierung, da während der Selektion auf den Agarplatten mittels Antibiotika (Ampicillin) resistente Klone isoliert werden. Nachzuweisen ist dies durch eine blaue Färbung, doch dazu später.
Bevor wir anfangen, müssen alle Reagenzien aufgetaut und durchmischt werden. Anschließend werden die Reagenzien sofort auf Eis gestellt.
Durchführung:
1) Schneiden des Plasmids pUCD mit den Restriktionsenzymen Bam H I und Hin d III
In dem ersten Teil des Versuchs wird das Plasmid pUCD mit den Restriktionsenzymen Bam HI und Hin D III geschnitten. Dies geschieht indem wir die Reagenzien entsprechend der angegebenen Mengen und Reihenfolge in das vorbereitete Eppendorf-Gefäß pipettieren. Gruppe 1 und 2 stellen folgenden Restriktionsansatz her: Dieser enthält 2 x 14 ml steriles Wasser; 4 ml Puffer B; 4 ml der Plasmid-pUCD-DNA; 2 ml des Restriktionsenzyms Bam H I und 2 ml des Restriktionsenzyms Hin dIII.
Der Restriktionsansatz der Gruppen 3 und 4 setzt sich nahezu genauso zusammen, einziger Unterschied ist, dass der Plasmidring pUCD-lacZ-DNA enthält. Dieses ist ein E. coli eigenes Gen. Um eine erfolgreiche Klonierung dieses Gens nachweisen zu können, wird im Experiment der E. coli Stamm K12 verwendet, dessen lacZ-Gen defekt ist.
Außerdem bewirkt das pUCD-lacZ-Plasmid bei Bakterien eine Ampicillinresistenz, was später von großer Bedeutung sein wird!
Für das Restriktionsenzym Bam H I ist in 5’- 3’- Richtung die Erkennungssequenz ...GGATCC... und in 3’- 5’- Richtung die Erkennungssequenz ...CCTAGG... charakteristisch. Für Hin d III sind es in 5’- 3’- Richtung ...AAGCTT... und in 3’- 5’- Richtung ...TTCGAA... Man spricht in diesem Fall aufgrund der besonderen Symmetrie von Palindromen.
Hier entstehen ebenfalls die überhängenden Enden oder sticky ends, die in der Gentechnik dazu dienen, fremde Genabschnitte unterschiedlichen Ursprungs in ein Plasmid einzubauen.
Wir mischen nun den Restriktionsansatz durch auf- und abpipettieren. Dann inkubieren wir diesen bei 37° C für 60 min. Um die Reaktion zu stoppen, inaktivieren wir das Restriktionsenzym durch fünfminütiges Erhitzen bei 65° C. Danach wird die Probe bis zur weiteren Anwendung auf Eis gelegt, oder bei –20 °C eingefroren.
Vorbereitung des Agarose-Gels
Um die Elektrophorese durchführen zu können, benötigt man zum einen den verdünnten TBE-Puffer. Dieser wird aus 30 ml Elektrophoresepuffer TBE (10fach konzentriert) und 270 ml destilliertes Wasser hergestellt.
Des Weiteren wird das Agarose-Gel benötigt, welches aus 30ml des verdünnten TBE-Puffers und 300 mg Agarose besteht. Durch kochen in der Mikrowelle wird die Agarose gelöst.
Nun folgt das gießen des Agarose-Gels. Hierzu wird die Elektrophoresekammer auf eine ebene Oberfläche gestellt. Nun wird der Gelträger eingesetzt und mit den Trennkeilen abgedichtet. Das Füllen des Gelträgers kann nun mit der Agaroselösung beginnen. Es ist darauf zu achten, dass die Lösung nicht mehr als 7-8 mm des Gelträgers bedeckt.
Als letztes werden die Probenkämme eingesetzt. Nach etwas 20 min ist das Gel erstarrt und die Keile sowie die Probekämme können vorsichtig entfernt werden.
Bevor die Proben auf das Agarose-Gel aufgetragen werden, wird der restliche TBE-Puffer in die Kammer gegossen, sodass das Gel ca. 1 mm mit TBE-Puffer bedeckt ist. Nun können die Proben aufgetragen werden.
2) Vorbereitung der Proben für die Elektrophorese
Die Gruppen 1 und 2 stellen zuerst einen DNA-Marker aus 8 ml DNA-Marker, 7 ml sterilem Wasser und 2 ml DNA-Auftragspuffer her. Zusätzlich wird eine Kontrolle aus ungeschnittener pUCD-DNA hergestellt. Diese besteht aus 2 ml ungeschnittener Plasmid-pUCD-DNA, 13 ml sterilem Wasser und 2 ml DNA-Auftragspuffer.
Der Restriktionsansatz wird durch 15 ml Restriktionsansatz und 2 ml DNA-Auftragspuffer in einem dritten Eppendorfgefäß verlängert.
Die Gruppen 3 und 4 stellen zeitgleich eine Kontrolle aus geschnittener pUCD-lacZ-DNA her.
Diese besteht aus 15 ml Restriktionsansatz und 2 ml DNA-Auftragspuffer.
Außerdem wird eine Kontrolle des lacZ-Genfragments angesetzt, die sich aus 10 ml
lacZ-Gen, 5 ml sterilem Wasser und 2 ml DNA–Auftragspuffer zusammensetzt.
Zuletzt wird nochmals ein DNA-Marker, wie bereits oben beschrieben, angesetzt. Die Proben werden bis zur weiteren Verwendung auf Eis aufbewahrt.
3) Elektrophorese
Nachdem die Elektrophoresekammer vorbereitet wurde, können wir nun die Proben in die Taschen des Agarosegels pipettieren. Es werden jeweils 10 ml der Proben benötigt.
Insgesamt hat jede der vier Gruppen jeweils 3 Spuren.
Gruppe 1 und 2:
Spur 1: 10 ml DNA- Marker
Spur 2: 10 ml Kontrolle 2 (ungeschnittene pUCD-DNA)
Spur 3: 10 ml Restriktionsansatz pUCD mit Bam H I und Hin d IIII ( R) verdaut
Gruppe 3 und 4:
Spur 4: 10 ml Restriktionsansatz pUCD-lacZ mit Bam H I und Hin dIII verdaut.
Spur 5: 10 ml Kontrolle 5 (lacZ-Gen)
Spur 6: 10 ml DNA- Marker III
Die Elektrophorese wird nun gestartet und muss beendet werden, bevor die Fragmente die zweite Geltaschenreihe erreichen (20 – 30 min.). Nach Beendigung wird eine blaue Färbelösung in eine Glas- oder Plastikschale gefüllt und anschließend vorsichtig das Agarose- Gel dazugegeben. Dieses lassen wir 15 Minuten in der Färbelösung und schütten dann die Färbelösung in die Flasche zurück, da diese wiederverwendbar ist.
Zum Entfärben des Gels spülen wir die Schale mehrmals mit destilliertem Wasser, bis nach 10 Minuten das Bandenmuster gut erkennbar ist.
4) Versuchsauswertung: Schneiden des Plasmids pUCD mit den Restriktionsenzymen
Bam H I und Hin d III

|
Spur 2 |
Spur 3 |
Spur 4 |
Spur 5 |
Spur 6 |
Die Fragmentgröße des Restriktionsansatzes wird mit Hilfe einer Eichkurve bestimmt. Die Werte stammen aus dem DNA-Marker.
|
Fragmentgröße |
log der |
Wanderungsstrecke |
|
in bp |
Fragmentgröße |
in mm |
|
4796* |
3,680879174 |
10 |
|
3530 |
3,547774705 |
10,5 |
|
2027 |
3,306853749 |
15 |
|
1904 |
3,279666944 |
16,5 |
|
1480* |
3,170555059 |
17,5 |
|
947 |
2,976349979 |
20,5 |
|
831 |
2,919601024 |
21 |
* Da die Wanderungsstrecke bei mehreren Fragmentgrößen identisch waren, wird der Mittelwert der Fragmentgrößen gemittelt

Mit Hilfe der Formel y= - 0,0631x + 4,2696, können wir nun die Fragmentgrößen unserer Ansätze bestimmen.
|
|
Wanderungsstrecken |
Log |
Fragmentgröße |
|
pUCD ungeschnitten |
16 |
3,26 |
1819 |
|
pUCD geschnitten |
14 |
3,3862 |
2433 |
|
pUCD-lacZ geschnitten |
14 |
3,3862 |
2433 |
|
pUCD-lacZ geschnitten |
12,5 |
3,48085 |
3025 |
|
lacZ-Gen |
12,5 |
3,48085 |
3025 |
Anhand der Ergebnisse erkennen wir, dass die Plasmide in den oben genannten Fragmentgrößen geschnitten worden sind.
Anhand der gegebenen Werte, können wir nun unsere Werte mit den in der Literatur gegebenen Werten vergleichen.
Im Vergleich mit den Literatur-Werten sind unsere Werte sehr gut. Messfehler bei der Wanderungsstrecke könnten dazu geführt haben, dass nicht alle Werte genau übereinstimmen.
|
|
Literatur Fragmentgrößen in bp |
Unsere Fragmentgröße in bp |
|
pUCD ungeschnitten |
2408 |
1819 |
|
pUCD geschnitten |
2378 |
2433 |
|
pUCD-lacZ geschnitten |
2378 |
2433 |
|
pUCD-lacZ geschnitten |
3291 |
3025 |
|
lacZ-Gen |
3291 |
3025 |
5) Ligation des lacZ-Gens mit dem linearisierten Plasmid pUCD
Wir bereiten den Ligationsansatz vor, indem wir 10 ml steriles Wasser, 2 ml Ligasepuffer, 4 ml lacZ- Gen, 2 ml Restriktionsansatz der Gruppe 1 + 2 und 2 ml DNA-Ligase in ein Eppendorfgefäß geben.
Als nächstes inkubieren wir den Ligationsansatz 5 Minuten bei Raumtemperatur. Dies ermöglicht die Ligationsreaktion.
Bei der Ligation kommt es zu einer kovalenten Bindung der Phosphatgruppe am 5´- Ende und der OH- Gruppe am 3´- Ende der Nukleotide.
6) Herstellung der Agarplatten
Agar ist ein Polysaccharid aus Rotalgen und bildet die Grundlage, in welcher die Nährstoffe gelöst werden.
Für die Vorbereitung der Nährböden wiegt eine kleine Gruppe 8 g LB-Agar ab und gibt diesen zusammen mit 220 ml destilliertem Wasser in ein Gefäß, das lose verschlossen wird.
Dies wird nun 20 min in einem Sterilisator sterilisiert. Dabei löst sich der Agar.
Nach dem Aufkochen wird das Gefäß in kurzen Abständen geschwenkt und danach in einem Wasserbad auf 50 °C abgekühlt, wobei darauf geachtet werden muss, dass der Agar nicht unter 45° C abkühlt, da er sonst vorzeitig erstarrt und damit wertlos wäre.
Ein erneutes Aufkochen würde den Nährboden jetzt nutzlos machen, da das Ampecillin zerstört würde.
Jede Gruppe erhält 2 Petrischalen mit LB-Agar, Ampicillin, IPTG und X-Gal, zudem werden 4 Kontrollschalen gegossen:
K1: LB, E.coli nichttransformiert
K2: LB/Amp/IPTG/X-Gal, E.coli nichttransformiert
K3: LB, E. coli mit pUCD-lacZ
K4: LB/Amp/IPTG/X-Gal, E. coli mit pUCD-lacZ
Außer in K1 und K3 ist also der LB-Agar mit Ampicillin, IPTG und X-Gal versetzt.
Der Agar wird dünnschichtig hineingegossen. Der Deckel sollte zur Bewahrung der Sterilität sofort wieder geschlossen werden. Mit dem verbliebenen Agar gießen wir noch weitere Platten als Reserve. Nach dem Erstarren des Agars werden die Petrischalen umgedreht, damit das sich bildende Kondenswasser nicht auf den Agar tropft und eventuelle Verunreinigungen verhindert werden.
Anschließend werden die Agarplatten in einem Kühlschrank bis zur weiteren Verwendung aufbewahrt.
Das Ampicillin in den Agarplatten dient der Selektion von Bakterien, die ein Plasmid aufgenommen haben.
7) Transformation und Selektion:
Transformation der Bakterien mit dem ligierten Plasmid pUCD
Jede Gruppe erhält nun ein Eppendorfgefäß mit 200 ml kompetenten E. coli K12 Zellen und lässt es etwa 15 Minuten auf Eis auftauen. Dann werden 20 ml des Ligationsansatzes (aus Punkt 5) hinzugefügt und 15 Minuten auf Eis inkubiert. Danach stellen wir den Transformationsansatz für genau 5 Minuten in ein 37 °C Wasserbad und daraufhin sofort für 5 Minuten wieder auf Eis.
Es werden 500 ml LB- Medium zu unserem Transformationsansatz gegeben. Damit sich die Zellen erholen können, inkubieren wir das Ganze wieder für 15 Minuten in einem 37 °C Wasserbad.
Da Bakterienzellwände für Fremd-DNA nicht durchlässig sind, wurden diese bei den E. coli K12 Zellen mit Calciumchlorid behandelt. Dadurch ist das Bakterium in der Lage für kurze Zeit ein Plasmid aufzunehmen. Solche Bakterien bezeichnet man als transformationskompetent.
8) Kontrollansätze
Zuerst verdünnen wir die pUCD-lacZ-Lösung für die Transformationskontrolle, während eine andere Gruppe ein Gefäß mit 200 ml kompetenten E. coli K12 für die Kontrollen K1 bis K4 ca. 15 Minuten auf Eis auftauen lässt.
Dann werden die aufgetauten kompetenten E. coli K 12 Zellen auf zwei Eppendorfgefäße à 100 ml aufgeteilt und eines der Gefäße durchläuft anschließend den Transformationsversuch, ohne dass die Zellen mit einem Plasmid transformiert werden (K1, K2). In das andere Gefäß (K3, K4) pipettieren wir 2 ml pUCD-lacZ, welches wir am Anfang verdünnt haben.
Anschließend stellen wir die Proben wieder für 5 Minuten auf Eis.
Nach den 5 Minuten geben wir je 250 ml LB- Medium zu unseren Ansätzen und inkubieren diese erneut für 15 Minuten in einem 37 °C Wasserbad.
9) Ausplattieren der Bakterien
- transformiert mit den ligierten Plasmiden
Jede Gruppe gibt auf die dafür vorbereitete Selektionsplatte einmal 40 ml und auf die andere Selektionsplatte 100 ml des eigenen Transformationsansatzes, den wir zuvor gut durchmischt haben, da Bakterien sich schnell absetzen. Die Bakteriensuspension wird gleichmäßig auf den Platten mit Hilfe eines Glasspatels, der zuvor durch eintauchen in Ethanol und das anschließende abflammen sterilisiert wurden, verteilt.
- der nichttransformierten E. coli K12
Aus dem Kontrollansatz K1/K2 werden jeweils 40 ml auf die Kontrollplatte K1 und 40 ml auf die Selektionsplatte K2 gegeben. Die Zellen werden, wie zuvor auch schon beschrieben, ausplattiert.
- transformiert mit pUCD-lacZ
Wir geben 40 ml des Kontrollansatzes K3/K4 auf die Kontrollplatte K3, sowie die gleiche Menge auf die Selektionsplatte K4 und plattieren wieder.
Dann müssen die Platten ca. 15 Minuten mit geschlossenem Deckel trocknen. Wenn dies geschehen ist, werden die Platten mit dem Boden nach oben inkubiert, sodass kein Kondenswasser auf die Agaroberfläche tropfen kann. Die Bakterien können nun über Nacht bei 37 °C wachsen.
10) Versuchsauswertung: Transformation und Klonierung
Ergebnisse:
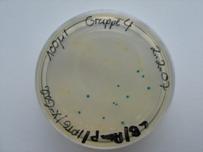
Probe 1 (40 ml): Es sind sowohl weiße als auch blaue Kolonien entstanden. Die weißen Kolonien sind durch die Religation zu erklären. Bei den blauen Kolonien hat das Bakterium das ligierte pUCD Plasmid mit dem lacZ-Gen aufgenommen.
Probe 2 (100 ml): Es sind dieselben Ergebnisse wie bei Probe 1 zu beobachten, jedoch sind die blauen Kolonien in größerer Zahl vorhanden, da wir eine größere Menge des Transformationsansatzes verwendet haben.
K1 + K2: Die Versuche dienen der Kontrolle, dass die Bakterien lebensfähig waren und dass die nichttransformierten Bakterienstämme nicht verunreinigt waren.
Auf den LB-Agarplatten findet ein Wachstum statt, da dort kein Ampicillin vorhanden ist, welches das Wachstum stoppen könnte. Auf den LB/Amp/IPTG/X-Gal-Agarplatten findet kein Wachstum statt, da keine Resistenzgene vorhanden sind.
K3 + K4: Auf den LB-Agarplatten ist ein Wachstum zu beobachten, da jedes Bakterium dort wachsen kann. Auf den LB/Amp/IPTG/X-Gal-Agarplatten wachsen blaue als auch weiße Kolonien, da die Bakterien das pUCD-lacZ-Gen (blaue Kolonien) enthalten bzw. durch Religation kein lacZ-Gen (weiße Kolonien) enthalten, wohl aber ein Ampicillinresistenzgen.

|
|
|
|
LB-Agarplatten |
|
LB/Amp/IPTG/X-Gal-Agarplatten |
|
|
|
|
|
|
|
|
|
|
|
|
|
Wachstum |
Kolonienzahl |
Wachstum |
Kolonienzahl |
|
|
|
|
+ / - |
weiß |
+ / - |
weiß / blau |
|
|
|
|
|
|
|
|
|
Probe 1 |
E.coli K12 transformiert mit |
|
|
+ |
300 / 10 |
|
|
|
ligirtem pUCD-lacZ-Plasmid |
|
|
|
|
|
|
Probe 2 |
aus dem Ligationsansatz |
|
|
+ |
420 / 11 |
|
|
|
|
|
|
|
|
|
|
K1 + K2 |
E.coli K12 nichttransformiert |
+ |
>1000 |
- |
0 |
|
|
|
|
|
|
|
|
|
|
K3 + K4 |
E.coli K12 transformiert mit |
+ |
>1000 |
+ |
400 / 56 |
|
|
|
pUCD-lacZ-Plasmid |
|
|
|
|
|
Durch das Transformationsexperiment haben sich die Bakterien, die einem weißen Klon entstammen, phänotypisch nicht verändert, genotypisch aber schon. Es wurde ein pUCD – Plasmid aufgenommen, die Bakterien besitzen das Gen für Ampicillin-Resistenz. Sie haben jedoch kein lacZ-Gen aufgenommen. Es kam zu einer Religation/ Einbau des zuvor herausgeschnittenen 30 bp- Fragments.
IPTG dient als Induktor für die Expression des lacZ-Gens. IPTG bindet an den Repressor des lac-Operons und inaktiviert diesen, sodass die lacZ-Strukturgene transkribiert werden können. Daraus folgt dann die Synthese der ß-Galactosidase.
Die Bakterien, die einem blauen Klon entstammen, haben ein blaues Erscheinungsbild, da die gebildete ß- Galactosidase das X-Gal in einen blauen Indigofarbstoff gespaltet hat.
Die genotypische Änderung ist folgende: Ein pUCD-lacZ-Plasmid wurde aufgenommen und das Gen für die Ampicillin-Resistenz ist vorhanden, außerdem ermöglicht das vorhandene lacZ-Gen die Synthese des Enzyms ß-Galactosidase.
Nach längerer Inkubation bei 37°C wachsen aus den blauen Kolonien auf der Platte weiße Bakterienkolonien an den Rändern nach. Das liegt daran, dass die Bakterien, die das pUCD- lacZ-Plasmid aufgenommen haben und es expremieren, ß-Lactamase bilden. Diese wird in das umgebende Medium abgegeben und zerstört das Ampicillin. Dort können dann auch Bakterien wachsen (weiße Kolonien), die kein Plasmid aufgenommen haben.
Es werden hinsichtlich des Genotyps an einem Sicherheitsstamm wie z.B. E. coli K12 Anforderungen gestellt. Diese sind, dass die Bakterien nicht pathogen sein dürfen und dass man Veränderungen im Erbgut bewirken muss, sodass der menschl. Darm nicht mehr besiedelt werden kann.
Das Sicherheitsplasmid wie es hier verwendet wurde (pUCD) ist auf das Bakterium E. coli beschränkt und kann auf natürliche Weise nicht in Bakterien übertragen werden. Die Gene tragen die Information für die Übertragung der Plasmide von Zelle zu Zelle. Diese wurden gezielt entfernt. Es wird kein Fremdgen eingebaut, sondern das lacZ-Gen, welches der Wildtyp besitzt und in die eingesetzten Bakterien eingeschleust wurde.
Bakterien schützen sich durch Methylasen davor, ihre eigene DNA enzymatisch zu zerschneiden. Diese binden gezielt an den Stellen, an denen Restriktionsenzyme binden.
11) Diskutieren der Versuchsergebnisse und Fazit :
Wie man nun in der Auswertung erkennen kann, haben wir bei Probe 1 (die transformierten E.coli Zellen mit dem legierten pUCD-lacZ-Plasmid) 300 weiße Kolonien und 10 blaue.
Dies ist ein nahezu optimales Ergebnis. Durch die ß-Galactosidase wurde der blaue Indigofarbstoff expremiert, wodurch deutlich erkennbar ist, dass das pUCD-lacZ-Plasmid erfolgreich in die E.coli-Zellen eingebaut wurde und folglich auch eine Ligation des LacZ-Gens stattgefunden hat.
Probe 2 hat ähnlich optimale Ergebnisse, da sie aus demselben Ligationsansatz stammt, nur in größerer Menge aufgetragen wurde (100 ml statt 40 ml).
Die Kontrollversuche 1+2 (die nicht transformierten E.coli Zellen) enthalten kein Amp-Resistenz-Gen. Auf den einfachen LB-Agarplatten können diese sich also zahlreich vermehren, auf den mit Ampicillin Agarplatten fand kein Wachstum statt, da unsere E.coli-Zellen kein Resistentzgen enthalten. Wie schon in den zu erwartenden Ergebnissen erwähnt, musste dies so sein, da es sich um einen Kontrollversuch handelte. Genauso war es auch bei den Kontrollversuchen 3+4, den transformierten Zellen. Diese wurden nun auf den einfachen LB-Agarplatten, sowie auf den LB/Amp/IPTG/X-Gal-Agarplatten aufgetragen. Bei beiden erkennt man starkes Wachstum.
Ein wohl optimales Ergebnis wird auf den LB/Amp/IPTG/X-Gal-Agarplatten erzielt. Dort haben wir nun 400 weiße Kolonien und 56 blaue. Bei unseren Versuchen hatten wir zwar nicht ganz so viele blaue Kolonien, aber dennoch waren diese ja erfolgreich verlaufen und solche Ergebnisse wie bei den Kontrollversuchen kann man bei einem solchen Schülerversuch nicht erzielen. Die Kontrollversuche dienen ja schließlich auch, wie der Name schon sagt, zur Kontrolle, als Richtlinie also. Wenn die Ergebnisse dieser Versuche anders ausgesehen hätten, wäre vermutlich bei der Herstellung der Agarplatten etwas schief gelaufen.
Abschließend lässt sich sagen, dass wir als Bio-Kurs bei diesem spannenden Versuch weitgehend alles richtig gemacht haben und mit unseren Ergebnissen auf jedenfall zufrieden sein können. Es hat Spaß gebracht und gleichzeitig die doch manchmal ermüdende Theorie spielerisch veranschaulicht und das ganze Thema schmackhaft gemacht.
Fazit: Daumen hoch Herr Kirchhoff
Protokollanten:
Jan-Frederik Schlie, Julius Dommer,
Wiebke Vorrath, Dorothee Sayk, Christin Bourjau und Claudia Vehlow